You read it right: PeekMed is now certified for European Union Medical Device regulation – EU MDR Directive, for short.
If you are not familiar with the topic, it's how medical devices are regulated in the European Union. While being certified for EU MDR Regulation is important for us, it also impacts you.
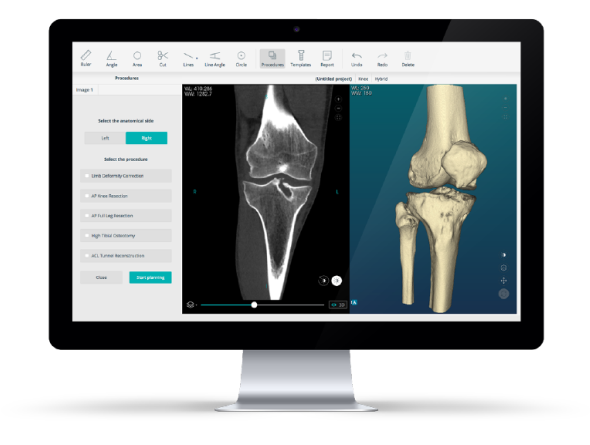
As of today, when you are planning surgeries with PeekMed, you can be sure that you are dealing with a high-quality system. PeekMed is not only foreseeing any issue that could happen but is also prepared to deal with the future.
This is a very short resume regarding the topic, however, if you would like to know more about it, please, keep reading.
-2.png?width=4800&name=PeekMed_EU_MDR%20(1)-2.png)
What does EU MDR mean in Medical terms?
EU MDR stands for European Medical Device Regulation. Introduced by European Commission, it is normally identified as Regulation (EU) 2017/745 (EU MDR). The regulation entered into application on 26 May 2021 in the European market.
The regulation is a set of compliant regulatory systems. These are mandatory to ensure the safety and performance of medical devices manufactured and sold in the EU. This is especially important to establish a robust, transparent, and predictable legal environment for medical devices.
MDR in Orthopedics: why does it affect you and PeekMed?
MDR comes to replace MDD. Compared to the former mandatory directive, MDR legal requirements are tighter than ever. This significantly improves the safety and performance of medical devices placed on the EU market, therefore the health of all EU citizens.
How does this impact you? We already knew that our system was qualified to provide the necessary support for clinical evaluation and add value to the supply chain. Now, you can be sure that by using our solution you are trusting the right partner.
It's not just us who are confident about PeekMed's high-quality system. The validation is, as we’ve pointed out, very tight and done by a third-party auditor – also referred to as the notified body.
PeekMed was already certified for MDD (often identified in technical documentation as CE marking). However, the requirements of this directive were not specifically tailored and updated to software, creating some gray areas in the law. MDR, for the first time, introduced the term Medical Device Software and extra requirements for it.
“MDR was a great achievement for us! It’s an extremely specific validation that our efforts to build an orthopedic device capable to provide the necessary clinical evidence to surgeons.”, explains Sara Silva, Co-founder and Chief Quality Officer of PeekMed.
“Before MDR, our enormous efforts to create and maintain a quality and safe medical device paid off huge! That ensured that our MDR compliance plan could run as smoothly as it did.”, Sara adds.
If you would like to have more information on what changed with this new MDR directive, please, follow along.
Unique Device Identification (UDI) and EUDAMED
Traceability was one of the major changes applied by the new directive. For that purpose, identification for all medical devices based on the Unique Device Identifier (UDI) system is now mandatory.
This system aims to uniquely identify each batch or individual medical device. Therefore it enhances the effectiveness of the activities related to the safety of the devices after being manufactured.
In other words, if a problem in a healthcare institution happens because of a medical device, it can be easily identified and traced.
As PeekMed is FDA Cleared since 2018, the UDI system was already implemented and functioning in the company, so compliance was easily achieved.
MDR introduced EUDAMED, a public database with information on all medical devices certified to be compliant and placed on the EU market.
The goal is to strengthen market surveillance and transparency in the medical device field providing competent national authorities with quick access to information.
EUDAMED is an ongoing process that will gain even more importance in the future. We are glad to already be on board with it.
Manufacturers of Class IIa Medical Device
We can proudly say that we are a compliant class IIa medical device manufacturer! The requirements to fit these criteria became much higher with MDR but we are achieving all of them.
Although these requirements required some work, they have strengthened the product and the company. We are even more confident that what we are doing is up to our customers' needs.
This topic continues with our quality management system (QMS) being certified according to EN ISO 13485 and complying with MDR, too.
Our QMS is provided with the necessary tools to help develop software capable of helping orthopedic surgeons and therefore thousands of patients. PeekMed is designed with quality in mind, so it can perform as you expect and need.
Post-Market Surveillance and Vigilance
Unlike its predecessor, the new MDR is less focused on the pre-approval stage of medical device manufacturing. Instead, it promotes a life-cycle approach to medical device regulation.
Regarding the safety of devices, after they are sold, many objective requirements were introduced by MDR.
These requirements include the need to have a system that not only reacts to incidents but proactively collects and reviews information about the device.
We are always gathering information regarding our clients’ experiences with PeekMed. By doing so we can detect software bugs, trends, or service errors quickly.
By these means, we can improve PeekMed every day and provide a more helpful product for the world’s health. Every feedback, good or bad, is important to us!
